Clinical Summary
A 56-year-old man had been living with a diagnosis of Amyotrophic Lateral Sclerosis (ALS) for three years one of the most feared neurological diagnoses a patient can receive. Progressive weakness, balance difficulties, and bilateral lower limb involvement had severely impacted his quality of life. But something didn't quite fit. A comprehensive repeat electrophysiological evaluation at NeuroMet Wellness Care & Diagnostics told a different story and changed his prognosis entirely.
Patient Presentation
A 56-year-old male presented to our clinic with a three-year history of progressive bilateral lower limb weakness, difficulty walking, and impaired balance. He reported moderate-to-severe difficulty standing and walking unassisted, with significant gait instability. He also had moderate weakness in his right upper limb. These symptoms had been gradually worsening over the three years since his original diagnosis.
He had been previously diagnosed with Amyotrophic Lateral Sclerosis (ALS) (a progressive and fatal motor neuron disease affecting both upper and lower motor neurons — the nerve cells controlling voluntary muscle movement) based on prior EMG reports that had documented fibrillations (abnormal spontaneous electrical discharges from muscle fibres that indicate dying or damaged lower motor neurons a hallmark finding of ALS). These findings had driven the ALS label for three years. No family history of motor neuron disease was noted.
The patient was understandably distressed ALS carries a median survival of 2–5 years and has no cure. He came to us seeking a second opinion and further clarity.
Clinical Examination
On neurological examination, the patient demonstrated predominant upper motor neuron (UMN) signs (signs pointing to damage in the brain and spinal cord pathways, rather than the peripheral nerves or muscles themselves):
- Spasticity (stiffness and increased muscle tone) in bilateral lower limbs
- Hyperreflexia (exaggerated deep tendon reflexes)
- Bilateral extensor plantar responses (Babinski's sign — an abnormal reflex indicating UMN pathway involvement)
- Significant gait impairment with balance difficulty
- Weakness predominantly in bilateral lower limbs, moderate weakness in right upper limb
Crucially, there was no clinical evidence of lower motor neuron (LMN) involvement (no visible muscle wasting, no fasciculations — the visible twitching under the skin that is a hallmark of ALS). Bulbar function (speech and swallowing) was reported as involved clinically, prompting specific evaluation of the masseter (jaw-closing) muscle during EMG.
Investigations & Findings
A comprehensive electrophysiological study was performed, covering all four limbs and the bulbar territory.
Nerve Conduction Studies (NCV) — All Four Limbs:
- Motor nerve studies: Bilateral median, ulnar (upper limbs), posterior tibial, and common peroneal nerves (lower limbs) — all within normal limits
- Sensory nerve studies: Bilateral median, ulnar, and sural nerves — all within normal limits
- F-wave studies: Normal across all tested nerves
- Interpretation: No evidence of peripheral neuropathy, demyelination, or axonal loss
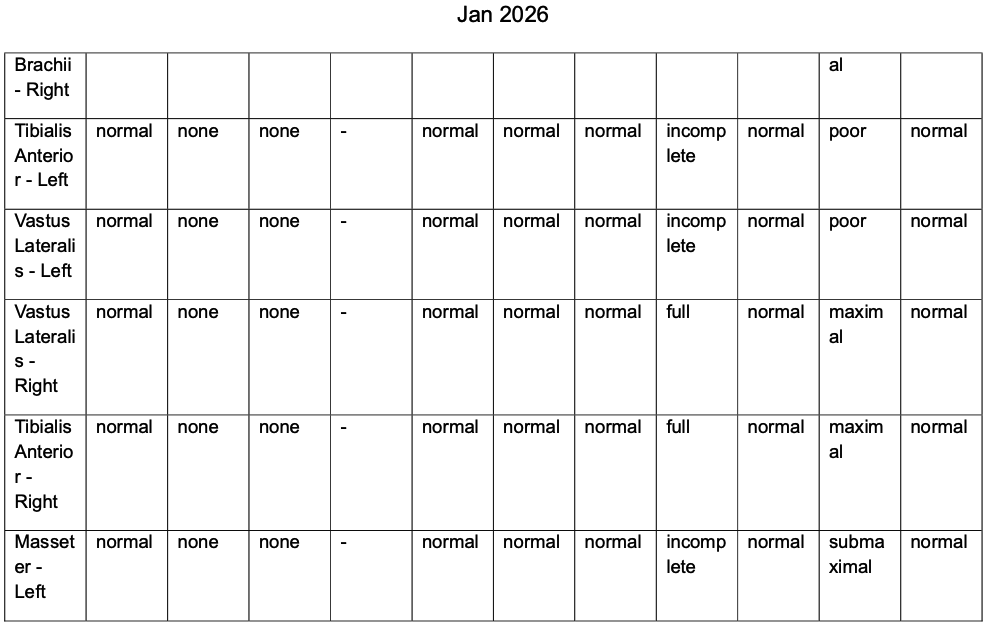
EMG (Needle Electromyography) — Comprehensive Muscle Sampling:
| Muscle | Spontaneous Activity | MUAPs | Classification |
|---|---|---|---|
| Flexor Pollicis Brevis (bilateral) | None | Normal | Normal |
| First Dorsal Interosseous (bilateral) | None | Normal | Normal |
| Abductor Pollicis Brevis (bilateral) | None | Normal | Normal |
| Biceps Brachii (bilateral) | None | Normal | Normal |
| Tibialis Anterior (bilateral) | None | Normal | Normal |
| Vastus Lateralis (bilateral) | None | Normal | Normal |
| Masseter — Left (Bulbar territory) | None | Normal | Normal |
There were no fibrillations, no fasciculations, and no positive sharp waves (the electrical signatures of dying or damaged lower motor neurons) in any muscle sampled across all four limbs and the bulbar region. MUAPs (Motor Unit Action Potentials — the electrical signals generated when muscle fibres contract) were normal in amplitude, duration, and configuration throughout.
The electrophysiology was unequivocally normal.
Diagnosis
Primary Lateral Sclerosis (PLS) — confirmed, with revision from prior ALS diagnosis.
Diagnostic reasoning:
The clinical picture — progressive spastic paraparesis, bilateral lower limb involvement with right upper limb weakness, hyperreflexia, and Babinski's sign over three years — pointed firmly to an upper motor neuron syndrome. The absence of any electrophysiological evidence of lower motor neuron involvement across a thorough multi-muscle EMG study, including the masseter for bulbar territory, ruled out ALS with confidence.
ALS requires lower motor neuron involvement (fibrillations, fasciculations, positive sharp waves, or neurogenic MUAPs) in at least two or more body regions alongside UMN signs — the so-called Gold Coast Criteria (the current international diagnostic standard for ALS). This patient met none of those LMN criteria.
PLS, in contrast, is a rare motor neuron disease affecting only the upper motor neurons — the pathways running from the brain down the spinal cord. It is far less aggressive than ALS, with most patients surviving for decades. While it does cause progressive disability, it is not the death sentence that ALS represents.
The prior EMG had documented fibrillations — which, in isolation and in the context of a clinically alarming presentation, had driven the ALS diagnosis. However, fibrillations are not exclusive to ALS and can be seen in a range of conditions including radiculopathy, myopathy, and even as technical artefact in suboptimal studies. The critical question was always whether a consistent, multi-muscle, multi-region LMN pattern was present — and on comprehensive repeat evaluation, it was not. This is a sobering reminder that a single EMG finding, without the full clinical-electrophysiological mosaic, can misdirect diagnosis with devastating consequences for the patient.
Treatment Approach
Following the revised diagnosis, management was restructured:
Edaravone (Aravon): The patient had been initiated on intravenous edaravone (a neuroprotective agent originally approved for ALS that reduces oxidative stress damage to motor neurons) for an initial 14-day induction cycle per protocol. He showed early functional improvement during this period. Given its neuroprotective mechanism, edaravone was continued as oral maintenance therapy.
Baclofen (10mg twice daily): A muscle relaxant targeting spasticity — the primary source of functional disability in PLS. This directly addresses the UMN-driven stiffness and improves mobility and comfort.
Physiotherapy — Structured Programme:
- Daily prolonged stretching (hip adductors, hamstrings, calf)
- Tone inhibition techniques
- Gait training with emphasis on symmetry
- Balance and postural control exercises
- Avoidance of aggressive strengthening and fatigue
- Assistive devices as needed
Supportive supplements: Coenzyme Q10, methylcobalamin (Vitamin B12), and a calcium-magnesium-DHA combination for neurological support.
Riluzole was not indicated given the revised PLS diagnosis, and was not prescribed.
Outcome & Follow-Up
The patient has shown functional stability on this regimen. Physiotherapy has been the cornerstone of maintaining independence and quality of life. He has been counselled transparently — PLS does not have a disease-modifying treatment, but it is fundamentally different from ALS in its trajectory. Most PLS patients do not require ventilatory support and survive for many years, often decades, albeit with progressive disability.
Prognosis counselling was conducted with the patient and family, emphasising that this is a manageable chronic condition, not a terminal one — a distinction that carries enormous psychological weight.
Follow-up was scheduled at four weeks.
Clinical Pearls
- ALS requires electrophysiological LMN evidence — without fibrillations, fasciculations, or neurogenic MUAPs in at least two regions, the diagnosis cannot stand. The Gold Coast Criteria exist for a reason.
- Bulbar EMG matters — masseter EMG is a critical and often overlooked step. In this case, a clean masseter trace helped exclude bulbar LMN involvement, sealing the PLS diagnosis.
- Fibrillations alone do not make ALS — the prior EMG had documented fibrillations, which triggered the ALS label. But fibrillations are non-specific and can occur in radiculopathy, myopathy, or suboptimal recordings. ALS requires a consistent multi-muscle, multi-region LMN pattern, not an isolated finding in one study.
- Repeat EMG is not redundant — a comprehensive reassessment covering multiple muscles across all regions including bulbar territory can be life-changing. In this case, it reversed a three-year-old fatal diagnosis.
- PLS vs ALS is not a semantic distinction — it is the difference between a palliative prognosis and a decades-long life. Getting this right matters profoundly to the patient and family.
This case was managed by Dr. Bhupesh Kumar Mansukhani, Neurologist & Director, NeuroMet Wellness Care and Diagnostics, Gurgaon. For appointments: www.neurometwellness.com
Patient details have been de-identified and shared with appropriate consent. This case study is for educational purposes only.
Specialty tag: Neurodiagnostics | Motor Neuron Disease
References
- Turner MR, et al. "Primary lateral sclerosis: consensus diagnostic criteria." Journal of Neurology, Neurosurgery & Psychiatry. 2020;91(4):373–377.
- Shefner JM, et al. "A proposal for new diagnostic criteria for ALS." Clinical Neurophysiology. 2020;131(8):1975–1978. [Gold Coast Criteria]
- Eisen A, et al. "Cortical influences drive ALS, while primary lateral sclerosis may be a disease of cortico-motoneurons." Brain. 2010;133(10):2843–2846.
- Gordon PH, et al. "Primary lateral sclerosis: an update." Amyotrophic Lateral Sclerosis. 2006;7(4):208–216.
- Writing Group; Edaravone (MCI-186) ALS 19 Study Group. "Safety and efficacy of edaravone in well-defined patients with amyotrophic lateral sclerosis." Lancet Neurology. 2017;16(7):505–512.